Single cell RNA sequencing (scRNAseq) analysis identifies the cell populations in the muscularis externa of the pig colon
Single cell RNA sequencing of the pig colon
Dataset Overview
Study Purpose: The advent of single-cell RNA sequencing (scRNA-seq) provides unprecedented opportunities for exploring gene expression profile at the single-cell level. Currently, scRNA-seq has become a favorable choice for studying the key biological questions of cell heterogeneity, since bulk RNA-seq mainly reflects the averaged gene expression across thousands of cells. The pig is a relevant experimental model due to its similarity to the human in nutrition, physiology and metabolic process, particularly in the enteric nervous system (ENS). However, up to date, no scRNA-seq protocol has been developed and applied on pig colon tissue. Here, we develop a protocol to prepare the cell suspensions from the muscularis externa including the myenteric plexus with a minimum of artificial induction in transcriptional changes during single-cell dissociation and faithfully investigate gene expression in specific cell types using 10x Genomics scRNA-seq.
Data Collection: The proximal (pC), transverse (tC) and distal colon (dC) were dissected from two adult Yucatan minipigs. The outer smooth muscle layers along with the myenteric plexus were peeled off from the underlying tissue using forceps. After cell suspension preparation, parts of cell suspensions from pC were subjected to RNA extraction, cDNA library construction and RNA sequencing on Hiseq3000 platform to verify the effects of Actinomycin D (ActD) application on transcriptional changes. After fluorescence-activated cell sorting (FACS) for isolation of live cells, cell suspensions from pC, tC and dC were used for 10x Genomics scRNA-seq on an Illumina NextSeq 500 Sequencer. The data were analyzed using Python 3.7.4 and R 3.6.2.
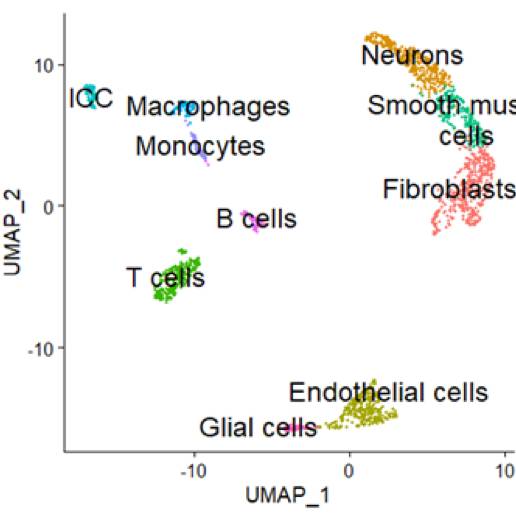
Primary Conclusion: (i) Immediate-early genes (IEGs) are rapidly and transiently induced by various cellular stimuli including synaptic activity. Therefore, they are good markers showing if the transcriptional profiling changes during single-cell dissociation. The sequencing data showed that there were 131 IEGs in pig transcriptome. Of them, 127 IEGs showed remarkably reduced induction with ActD application. This means that ActD application minimizes artificially induced transcriptional changes during single-cell dissociation and thus enables faithful characterization of baseline transcriptional profiles; (ii) The single-cell data were obtained from 2,091 cells in pC, 1,903 cells in tC and 3,918 cells in dC, sequenced to a depth of about 1,400 median genes per cell. Principal component analysis (PCA) was first carried out to reduce dimensionality. Based on the PCA output, non-linear dimensional reduction techniques, such as UMAP, were used to visualize and explore the datasets. Each point in UMAP depicts a single cell, colored according to cluster designation. There were 10 distinct major cell clusters found in muscularis externa of pC, tC and dC. The clusters in all three segments comprised fibroblasts (Fibro), neurons(Neu), endothelial cells (Endo), T cells (T), smooth muscle cells (Muscle), interstitial cells of Cajal (ICC), macrophages (Macro), monocytes (Mono), B cells (B) and glial cells (Glial).
Curator's Notes
Experimental Design: The full-thickness tissue samples of proximal (pC), transverse (tC) and distal (dC) colon were obtained from the pigs undergoing resection surgery. The samples were then cut into small pieces < 1 mm and tissue pieces were dissociated. The viable cells were collected via fluorescence-activated cell sorting (FACS), whose viability was assessed using a Countess II FL Automated Cell Counter. ~10,000 single cells per cell suspension were loaded on a Chromium Controller (10× Genomics) to generate single-cell gel beads in emulsion (GEMs). A Chromium Single Cell 3′ Reagent kit, v2 Chemistry (10× Genomics) was used to prepare single-cell RNA sequencing (scRNA-Seq) following the manufacturer's instructions. The libraries were sequenced on an Illumina NextSeq 500 Sequencer as 1 x 75 single-end reads. Cell Ranger v.1.3 (10× Genomics) was used to generate FASTQ files, which were then aligned to sus_scrofa genome and transcriptome using the Cell Ranger v.1.3 pipeline to generate ‘gene vs cell’ expression matrix. For filtering and unsupervised clustering, the gene expression matrix from Cell Ranger was used for downstream analysis (using Python 3.7.4 and R 3.6.2).
Completeness: This dataset is a part of a larger study pertaining to nerve stimulation on colonic motility in Yucatan minipigs.
Subjects & Samples: Young adult male (n=3) Yucatan pigs (RRID:NSRRC_0012), were used for this study.
Primary vs derivative data: Primary data folder is organized by subject and sample name and contains raw sequencing reads from NextSeq 500 1x75 High Output. For FASTQ generation, Illumina basecall files (*.bcl) were converted to FASTQs using Cell Ranger (10× Genomics), which uses bcl2fastq. FASTQ files were then aligned to pig genome and transcriptome using the Cell Ranger pipeline, which generates a ‘gene vs cell’ file containing barcoding, gene-expression, and expression matrix. The derivative folder contains filtered feature files ( barcodes, features, gene matrix) for each analyzed sample.
Important Notes: This dataset is currently undergoing image registration and will be updated once this process is complete.
Files
1 - 0 of 0 files
About this dataset
Publishing history
Cite this dataset
Tags
References
Is Supplemented by
Li, T., Yuan, P.-Q., & Tache, Y. (2020). A single cell RNA sequencing protocol for the pig colon v1. https://doi.org/10.17504/protocols.io.bgdmjs46

Copyright © 2026 University of Pennsylvania. All rights reserved.