Spatially tracked single-cell-scale RNAseq of porcine right atrial ganglionic plexus (RAGP) neurons
Neuronal samples from the porcine RAGP were collected through laser capture microdissection and run through single-cell-scale RNAseq
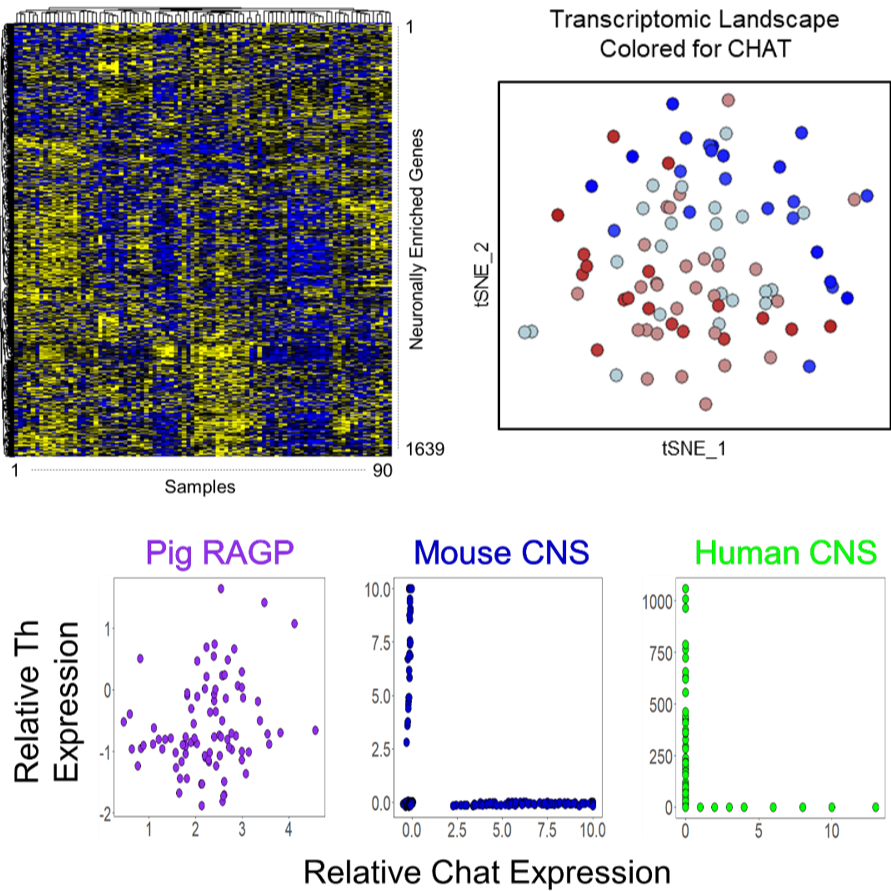
Dataset Overview
Study Purpose: The purpose of this study was to create a comprehensive atlas of the cardiac ICN in porcine RAGP at a cellular level providing gene expression profiles of cardiac neurons on a single-cell resolution. We developed an approach to appreciate the 3D organization of the intracardiac neurons, ICN, while at the same time permitting single-cell transcriptomics and connectomics.
Data Collection: This dataset contains results of single-cell-scale RNAseq run on 142 spatially tracked regional neuronal samples collected through Laser Capture Microdissection (LCM). After QC, 90 samples remain with detectable expression of over 15,000 genes, yielding 1,350,000 datapoints. These single-cell-scale samples were processed through RNAseq using a novel 3seq protocol, specifically designed for LCM samples (Foley et al. 2019). Fastq and bam files are included for all 142 samples that were assayed as well as raw counts and normalized counts for the 90 samples that passed QC.
Primary Conclusion: A novel protocol was used to perform single-cell-scale RNAseq on LCM samples, allowing, for the first time, spatially tracked single-cell-scale RNAseq. Imaging of these samples as they were collected through LCM allowed spatial tracking of these samples within their 3D context, allowing the transcriptional profiles of these neurons to be mapped back to their anatomical locations.
Curator's Notes
Experimental Design: The porcine heart was injected with FastBlue, a retrograde neuronal tracer, into the SAN before sacrifice, allowing the distinction between SAN-connecting neurons and non SAN-connecting neurons based on whether they stain for just cresyl violet (non SAN-connecting), or cresyl violet and FastBlue (SAN-connecting). Images were taken of the section as well as at several different levels of magnification both before the samples were lifted and after. Such sample tracking later allowed for mapping of the transcriptomic results back to their 3D locations. The individual neurons were then run through RNAseq using a novel 3seq protocol tailored for Laser Capture Microdissection. Out of 142 samples, 52 Samples with non-zero gene counts <6000 were considered as outliers. Additionally, 10800 genes that are present in very low quantities (<30 non-zero gene counts) were filtered out. A regularized log transformation was carried out using DESeq258. Researchers normalized the filtered data using a quantile regression method SCnorm59. The final matrix consisted of 90 samples and 15000 genes.
Completeness: This dataset is a part of a larger study " Spatially Tracked Single Neuron Anatomical and Molecular Profiling of the Pig Right Atrial Ganglionic Plexus"
Subjects & Samples: A total of 142 tissue samples were extracted from 1 young, female, Yucatan pig.
Primary vs derivative data: Raw sequencing data (Illumina sequencer’s base call files, BCLs) was converted to Fastq files using the Illumina bcl2fastq program and is available in the source data folder. Sequence alignment data can be found in the primary data folder as BAM files. A modified version of Sus_scrofa.Sscrofa11.1.95.gtf was used as a reference transcriptome. A list of neuronal genes enriched in the brain and neuronal tissues using data from the Genotype Tissue Expression (GTEx) project can be found in the derivative folder as gene counts in a text file format.
Files
1 - 0 of 0 files
About this dataset
Publishing history
Cite this dataset
Tags
References
Is Supplemented by
Kuttippurathu, L., Moss, A., & Vadigepalli, R. (2021). Single Cell scale RNA-seq Analysis Protocol to analyze Smart-3SEQ data from RAGP neurons of pig heart. https://doi.org/10.21203/rs.3.pex-962/v1
Described by
Moss, A., Robbins, S., Achanta, S., Kuttippurathu, L., Turick, S., Nieves, S., Hanna, P., Smith, E. H., Hoover, D. B., Chen, J., Cheng, Z. (., Ardell, J. L., Shivkumar, K., Schwaber, J. S., & Vadigepalli, R. (2020). A spatially-tracked single cell transcriptomics map of neuronal networks in the intrinsic cardiac nervous system. https://doi.org/10.1101/2020.07.29.227090

Copyright © 2026 University of Pennsylvania. All rights reserved.